Friday 19 April 2024
")
")



Le calcul le plus simple que l’on peut effectuer avec le code CRYSTAL est un calcul d'énergie en un seul point ( SPE - single point energy calculation). Il consiste à calculer la fonction d'onde et l'énergie pour un système cristallin donné avec une structure géométrique bien spécifiée, c'est-à-dire en un seul point fixe sur la surface d'énergie potentielle. L'énergie calculée est l'énergie totale, somme de l'énergie électronique et de l'énergie de répulsion nucléaire.
Pour effectuer un calcul SPE, la méthode théorique et l’ensemble de bases de la fonction d’onde doivent être spécifiés. L'analyse de la fonction d'onde calculée, une étape importante dans la prédiction des propriétés.
De nombreux aspects du comportement cristallin peuvent être compris lorsque l'énergie totale du système est connue, ou plus souvent, lorsque la différence d'énergie entre deux ou plusieurs configurations électroniques ou nucléaires est connue en fonction de certains paramètres. D'autres propriétés importantes sont liées aux dérivées de l'énergie totale. Par exemple, les dérivées premières par rapport aux coordonnées atomiques nucléaires donnent les forces agissant sur les atomes et permettent d'obtenir la géométrie d'équilibre lorsqu'une optimisation de la géométrie est effectuée. D'autres propriétés importantes sont liées aux dérivées secondes de l'énergie totale du système par rapport, par exemple, au volume, aux paramètres de la cellule unitaire et aux coordonnées fractionnaires des atomes. Les observables associés sont respectivement le module de masse, les constantes élastiques et les propriétés vibrationnelles.
Nous nous concentrerons principalement sur le calcul de l'énergie totale et sur quelques différences d'énergie associées pour les systèmes : MgO, Si et Be. Ce sont des exemples simples des catégories les plus importantes de composés solides : ionique, covalent et métallique, respectivement.
Un tutoriel sur le calcul de l'énergie et des propriétés d'un cristal ionique, MgO, est donné. Vous devez utiliser la même stratégie pour étudier un système covalent, le Silicium et un solide métallique le Béryllium.
Le code CRYSTAL requiert un fichier d’entrée (INPUT) structuré en trois blocs (données obligatoires) :
| Title | |
| input block 1 |
Geometry input (see tutorials: geometry editing) standard geometry input optional geometry optimization and editing keywords END |
| input block 2 |
Basis set input (see tutorial: basis set) standard basis set input optional basis set related keywords END |
| input block 3 |
Single particle Hamiltonian (default: RHF) and SCF control (see tutorial: Hamiltonian, computational parameters and SCF) SHRINK sampling in reciprocal space (for 1D-2D-3D systems only) optional general information and SCF related keywords END |
Le fichier INPUT doit toujours commencer par un titre par exemple :
MGO BULK
Block 1: Geometry
Une structure cristalline est déterminée par :
Ces informations peuvent être trouvées dans des bases de données cristallographiques, par exemple dans la base de données COD. En recherchant la structure MgO, la sortie de la base de données est la suivante (les informations nécessaires pour définir la structure cristalline sont mises en évidence) :
|
COL ICSD Collection Code 9863 DATE Recorded Jan 1, 1980; updated Jan 19, 1999 |
| NAME Magnesium oxide (Title) |
|
MINR Periclase FORM Mg O = Mg O TITL X-ray determination of electron-density distributions in oxides, Mg O, Mn O, Co O, and Ni O, and atomic scattering factors of their constituent atoms REF Proceedings of the Japan Academy PJACA 55 (1979) 43-48 AUT Sasaki S, FujinoK, TakeuchiY SYM x, y, z y, z, x z, x, y x, z, y y, x, z z, y, x x, -y, -z y, -z, -x z, -x, -y x, -z, -y y, -x, -z z, -y, -x -x, y, -z -y, z, -x -z, x, -y -x, z, -y -y, x, -z -z, y, -x -x, -y, z -y, -z, x -z, -x, y -x, -z, y -y, -x, z -z, -y, x -x, -y, -z -y, -z, -x -z, -x, -y -x, -z, -y -y, -x, -z -z, -y, -x -x, y, z -y, z, x -z, x, y -x, z, y -y, x, z -z, y, x x, -y, z y, -z, x z, -x, y x, -z, y y, -x, z z, -y, x x, y, -z y, z, -x z, x, -y x, z, -y y, x, -z z, y, -x |
| CELL a=4.217(1) b=4.217(1) c=4.217(1) alpha=90.0 beta=90.0 gamma=90. (Cell parameters) |
| V=75.0 D=3.56 Z=4 (Cell Volume and Density in gr/cm3) |
| SGR F m -3 m (225) - cubic (Hermann-Mauguin symbol and Space group number) |
|
CLAS m-3m (Hermann-Mauguin) - Oh (Schoenflies) PRS cF8 ANX AX PARM Atom__No OxStat Wyck -----X----- -----Y----- -----Z----- -SOF- |
|
Mg 1 2.000 4a 0. 0. 0. (Atom fractional coordinates) O 1 -2.000 4b 0.5 0.5 0.5 |
|
WYCK b a ITF Mg 1 B=0.312 ITF O 1 B=0.362 REM M PDF 43-1022 RVAL 0.013 |
Le bloc géométrie pour MgO s’écrira alors :
MGO BULK CRYSTAL 0 0 0 225 4.217 2 12 0. 0. 0. 8 0.5 0.5 0.5 |
Title of the job Dimensionality of the system (CRYSTAL => 3D) Crystallographic information (3D only) Space Group number Lattice parameters (minimal set of data) Number of atoms in asymmetric unit Atomic position specification in fractionary coordinates |
| TEST | Optional keyword to stop execution after geometry input |
| END | End of the geometry input section |
Exercice :
crystal < mgO_geom.d12 > mgo_geom.out
******************************************************************************* LATTICE PARAMETERS (ANGSTROMS AND DEGREES) - BOHR = 0.5291772083 ANGSTROM PRIMITIVE CELL - CENTRING CODE 5/0 VOLUME= 18.747822 - DENSITY 3.541 g/cm^3 A B C ALPHA BETA GAMMA 2.98186930 2.98186930 2.98186930 60.000000 60.000000 60.000000 ******************************************************************************* ATOMS IN THE ASYMMETRIC UNIT 2 - ATOMS IN THE UNIT CELL: 2 ATOM X/A Y/B Z/C ******************************************************************************* 1 T 12 MG 0.000000000000E+00 0.000000000000E+00 0.000000000000E+00 2 T 8 O -5.000000000000E-01 -5.000000000000E-01 -5.000000000000E-01 TRANSFORMATION MATRIX PRIMITIVE-CRYSTALLOGRAPHIC CELL -1.0000 1.0000 1.0000 1.0000 -1.0000 1.0000 1.0000 1.0000 -1.0000 ******************************************************************************* CRYSTALLOGRAPHIC CELL (VOLUME= 74.99128631) A B C ALPHA BETA GAMMA 4.21700000 4.21700000 4.21700000 90.000000 90.000000 90.000000 COORDINATES IN THE CRYSTALLOGRAPHIC CELL ATOM X/A Y/B Z/C ******************************************************************************* 1 T 12 MG 0.000000000000E+00 0.000000000000E+00 0.000000000000E+00 2 T 8 O -5.000000000000E-01 -5.000000000000E-01 -5.000000000000E-01 |
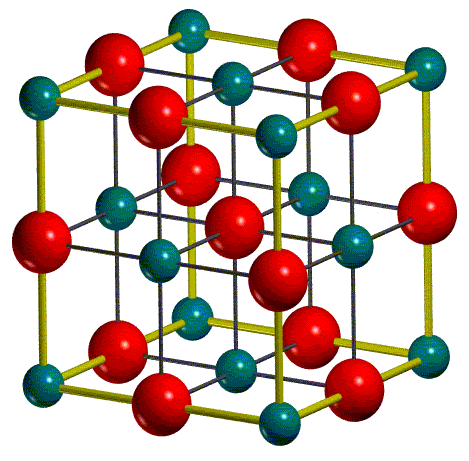
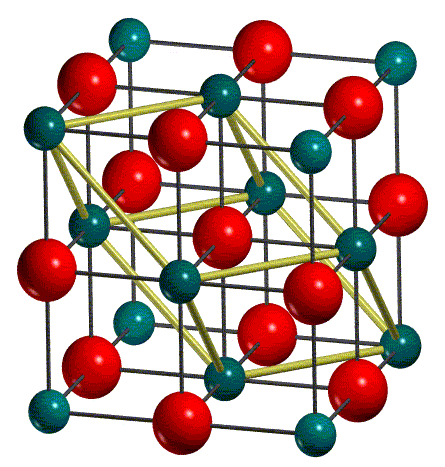
La maille conventionnelle est utilisée comme option standard en entrée. Notez que, pour une efficacité de calcul, CRYSTAL travaille sur la cellule primitive. Ainsi, la maille conventionnelle est transformée en maille primitive (1/4 de la maille conventionnelle pour les réseaux cubiques à faces centrées). Toutes les informations structurelles suivantes se réfèrent à la maille primitive.
Dans le fichier de sortie, les paramètres de maille de la maille primitive et les positions atomiques correspondantes (en unités fractionnaires) sont rapportés. Pour MgO, seulement deux atomes sont utilisés pour construire la maille primitive.
Dans la figure ci-dessous, représente la maille conventionnelle et la maille primitive du MgO :
| CONVENTIONAL CELL | PRIMITIVE CELL |
 |
 |
Block 2: Basis set
CRYSTAL effectue des calculs ab initio sur des systèmes périodiquesen utilisant une combinaison linéaire de fonctions de Bloch, définies en termes de fonctions locales, ci-après désignées par des orbitales atomiques (AO). Ces fonctions locales sont exprimées comme une combinaison linéaire d’un certain nombre de fonctions de type gaussien (GTF). Elle sont caractérisées par le même centre avec des coefficients fixes et des exposants définis dans l’entrée. Les OA appartenant à un atome donné sont regroupés en couches ( shells) s, sp, p, d, f.
|
shell type code |
shell type | AO | AO order | max shell charge |
|
0 |
s | 1 | s | 2 |
|
1 |
sp | 4 | s, x, y, z | 8 (s2 p6) |
|
2 |
p | 3 | x, y, z | 6 |
|
3 |
d | 5 | 2z2-x2-y2, xz, yz, x2-y2, xy | 10 |
|
4 |
f | 7 | (2z2-3x2-3y2)z, (4z2-x2-y2)x, (4z2-x2-y2)y, (x2-y2)z, xyz, (x2-3y2)x, (3 x2-y2)y | 0 - polarization only |
Pour chaque type d’atome il faut préciser :
le numéro atomique et le nombre de couches ns de l'ensemble de base atomique.
Pour chaque couche (ns) :
le type de jeu de base (0-1-2), le type de la couche (0-1-2-3-4), le nombre de gaussiennes primitives GTF ng, la charge électronique de la couche et le facteur d'échelle.
Pour chaque gaussienne primitive (ng - facultatif - à définir que pour le jeu de base de type 0) :
l’exposant et le coefficient de contraction.
Le nombre d'électrons attribués à chaque atome est la somme des électrons de chaque couche, comme indiqué en entrée. Cela a deux implications :
CRYSTAL peut utiliser des ensembles de bases généraux, y compris les fonctions s, p, d, f (polarisation uniquement) ou des ensembles de base Pople standard (stockés en interne).
|
BS input code |
description |
|
0 |
general basis set; gaussians exponent and contraction coefficients defined in input |
|
1 |
Pople STO-nG type basis set - internally stored |
|
2 |
Pople 3(6)-21G type basis set- internally stored |
Nous rapportons ci-dessous les différents ensembles de base pour le silicium :
|
STO-3G |
6-21G modified | 3-21G modified+polarization | free basis set |
14 3 1 0 3 2. 0. 1 1 3 8. 0. 1 1 3 4. 0. |
14 4 2 0 6 2. 1. 2 1 6 8. 1. 2 1 2 4. 1. 0 1 1 0. 1. 0.16 1. 1. |
14 5 2 0 3 2. 1. 2 1 3 8. 1. 2 1 2 4. 1. 0 1 1 0. 1. 0.16 1. 1. 0 3 1 0. 1. 0.5 1. |
14 4
0 0 6 2. 1.
16115.9 0.00195948
2425.58 0.0149288
553.867 0.0728478
156.340 0.24613
50.0683 0.485914
17.0178 0.325002
0 1 6 8. 1.
292.718 -0.00278094 0.00443826
69.8731 -0.0357146 0.0326679
22.3363 -0.114985 0.134721
8.15039 0.0935634 0.328678
3.13458 0.603017 0.449640
1.22543 0.418959 0.261372
0 1 2 4. 1.
1.07913 -0.376108 0.0671030
0.302422 1.25165 0.956883
0 1 1 0. 1.
0.123 1. 1.
|
| Number of shells: 3 | Number of shells: 4 | Number of shells: 5 | Number of shells: 4 |
| Number of AO: 9 | Number of AO: 13 | Number of AO: 18 | Number of AO: 13 |
| # electrons:14 | # electrons:14 | # electrons:14 | # electrons:14 |
La définition du jeu de base pour tous les atomes doit se terminer par (obligatoires) :
99 0 END |
Exercice :
crystal < mgO.d12 > mgo.out pour l'entrée mgo.d12 (géométrie + ensemble de base) avec les ensembles de base suivants :
Magnesium - ionic configuration,
Mg 2+: 1s(2) 2sp(8)
12 3
0 0 8 2. 1.
68371.875 0.0002226
9699.34009 0.0018982
2041.176786 0.0110451
529.862906 0.0500627
159.186000 0.169123
54.6848 0.367031
21.2357 0.400410
8.74604 0.14987
0 1 6 8. 1.
156.795 -0.00624 0.00772
31.0339 -0.07882 0.06427
9.6453 -0.07992 0.2104
3.7109 0.29063 0.34314
1.61164 0.57164 0.3735
0.64294 0.30664 0.23286
0 1 1 0. 1. 3sp shell charge 0.
0.4 1. 1.
Oxygen - ionic configuration, O2-: 1s(2) 2sp(8)
8 3
0 0 8 2. 1.
4000. 0.00144
1355.58 0.00764
248.545 0.05370
69.5339 0.16818
23.8868 0.36039
9.27593 0.38612
3.82034 0.14712
1.23514 0.07105
0 1 5 8. 1. 2sp shell charge 8.
52.1878 -0.00873 0.00922
10.3293 -0.08979 0.07068
3.21034 -0.04079 0.2043
1.23514 0.37666 0.34958
0.536420 0.42248 0.27774
0 1 1 0. 1.
0.210000 1. 1.
|
Magnesium - neutral configuration:
1s(2) 2sp(8) 3sp(2)
12 3
0 0 8 2. 1.
68371.875 0.0002226
9699.34009 0.0018982
2041.176786 0.0110451
529.862906 0.0500627
159.186000 0.169123
54.6848 0.367031
21.2357 0.400410
8.74604 0.14987
0 1 6 8. 1.
156.795 -0.00624 0.00772
31.0339 -0.07882 0.06427
9.6453 -0.07992 0.2104
3.7109 0.29063 0.34314
1.61164 0.57164 0.3735
0.64294 0.30664 0.23286
0 1 1 2. 1. 3sp shell charge 2.
0.4 1. 1.
Oxygen neutral configuration: 1s(2) 2sp(6)
8 3
0 0 8 2. 1.
4000. 0.00144
1355.58 0.00764
248.545 0.05370
69.5339 0.16818
23.8868 0.36039
9.27593 0.38612
3.82034 0.14712
1.23514 0.07105
0 1 5 6. 1. 2sp shell charge 6.
52.1878 -0.00873 0.00922
10.3293 -0.08979 0.07068
3.21034 -0.04079 0.20433
1.23514 0.37666 0.34958
0.536420 0.42248 0.27774
0 1 1 0. 1.
0.210000 1. 1.
|
Block 3: Hamiltonian and SCF computational parameters
Les hamiltoniens implémentés dans CRYSTAL sont HF (Hartree-Fock) et DFT (Density Functional Theory). Le choix par défaut est RHF (Hartree Fock). Tous les mots clés du troisième bloc d'entrée sont facultatifs, à l'exception de SHRINK et du mot clé END, qui doivent être insérés pour fermer la section.
Les choix de base pour l'hamiltonien sont reportés dans le tableau suivant :
|
Wavefunction |
HF | DFT | Characteristics |
|
Closed |
default | DFT | one determinant; |
|
Open |
UHF | SPIN | two determinants; one for alpha and one for beta electrons |
Le mot clé SHRINK doit être suivi des facteurs de contraction IS et ISP. IS est utilisé pour générer une grille commensurable de k-points dans l'espace réciproque, selon la méthode Pack-Monkhorst. La matrice hamiltonienne calculée dans l'espace direct est transformée de Fourier pour chaque valeur de k et diagonalisée pour obtenir des vecteurs propres et des valeurs propres. Le deuxième facteur de contraction, ISP, définit l'échantillonnage de k points utilisés pour la détermination de l'énergie de Fermi. Pour les systèmes conducteurs, l'ISP doit être d'au moins 2*IS.
D’autres mots clés importants mais pas nécessaire sont :
|
TOLDEE ite |
keyword SCF stops when absolute value of energy difference is less than 10-ite [default value is 6] |
|
MAXCYCLE imax |
keyword SCF stops when the number of cycles exceeds imax [default value is 50]. |
Exercice : (SPE - single point energy calculation)
crystal < mgO.d12 > mgo.out)un calcul SPE sur MgO avec les ensembles de base pour Mg et O utilisés auparavant. Utilisez les spécifications suivantes : HF hamiltonien (choix par défaut).SHRINK 4 4 FMIXING 30
|
Shrinking factor |
Number of k points | SCF energy (hartree) |
4. Recommencer le calcul en utilisant les spécifications suivantes : HF hamiltonien, SHRINK 8 8, FMIXING 30, TOLDEE 7 (valeur par défaut 6). Combien de cycles SCF pour atteindre la convergence ? Comment l'énergie du système a-t-elle été affectée par l'augmentation de la tolérance sur l'énergie ?
5. Répéter le calcul précédent avec TOLDEE 8. Combien de cycles SCF sont maintenant nécessaires pour atteindre la convergence ? Comment l'énergie du système est-elle affectée par l'augmentation de la tolérance sur l'énergie ?
6. Recueillez les résultats des 3 derniers exercices (calculs HF avec facteur de contraction 8 8) et remplissez le tableau suivant :
|
TOLDEE |
SCF energy (hartree) |
| 6 (default) | |
La détermination de la structure d'équilibre est de première importance dans la modélisation des systèmes chimiques. Le code CRYSTAL peut calculer des gradients analytiques de l'énergie par rapport aux paramètres de maille et aux coordonnées atomiques. L'optimisation de la géométrie peut être effectuée en coordonnées fractionnaires symétrisées ou en coordonnées internes redondantes.
Le mot clé OPTGEOM ouvre l'entrée d'optimisation de la structure (fermée par END[OPT]). Le mot-clé doit être inséré dans la section d'entrée de la géométrie (bloc 1) comme dernier mot-clé de la section, avant END[GEOM].
Par défaut, seule la relaxation des coordonnées atomiques est effectuée. Lorsque la position de tous les atomes est définie par la symétrie (comme dans le cas du MgO), aucune optimisation des coordonnées atomiques ne peut être effectuée.
Il est également possible d’optimiser les paramètres de la maille à des positions atomiques fixes avec le mot-clé CELLONLY. Les distorsions élastiques symétrisées sont générées en fonction de la symétrie du groupe d’espace. Ils ne coïncident pas avec la déformation des axes cristallographiques.
Le choix optionnel (INTREDUN) est l’optimisation des coordonnées internes redondantes des positions atomiques et des paramètres de maille.
Exercice : MgO - optimisation des paramètres de maille
MGO BULK - CELL OPT CRYSTAL 0 0 0 225 4.217 2 12 0. 0. 0. 8 0.5 0.5 0.5 OPTGEOM CELLONLY END END 12 3 0 0 8 2. 1. 68371.875 0.0002226 9699.34009 0.0018982 2041.176786 0.0110451 529.862906 0.0500627 159.186000 0.169123 54.6848 0.367031 21.2357 0.400410 8.74604 0.14987 0 1 6 8. 1. 156.795 -0.00624 0.00772 31.0339 -0.07882 0.06427 9.6453 -0.07992 0.2104 3.7109 0.29063 0.34314 1.61164 0.57164 0.3735 0.64294 0.30664 0.23286 0 1 1 0. 1. 0.4 1. 1. 8 3 0 0 8 2. 1. 4000. 0.00144 1355.58 0.00764 248.545 0.05370 69.5339 0.16818 23.8868 0.36039 9.27593 0.38612 3.82034 0.14712 1.23514 0.07105 0 1 5 8. 1. 52.1878 -0.00873 0.00922 10.3293 -0.08979 0.07068 3.21034 -0.04079 0.20433 1.23514 0.37666 0.34958 0.536420 0.42248 0.27774 0 1 1 0. 1. 0.210000 1. 1. 99 0 END SHRINK 8 8 FMIXING 30 END |
*******************************************************************************
CRYSTALLOGRAPHIC CELL (VOLUME= 73.60933348)
A B C ALPHA BETA GAMMA
4.19093535 4.19093535 4.19093535 90.000000 90.000000 90.000000
COORDINATES IN THE CRYSTALLOGRAPHIC CELL
ATOM X/A Y/B Z/C
*******************************************************************************
1 T 12 MG 0.000000000000E+00 0.000000000000E+00 0.000000000000E+00
2 T 8 O -5.000000000000E-01 5.000000000000E-01 5.000000000000E-01
|
******************************************************************
* OPT END - CONVERGED * E(AU): -2.746642641310E+02 POINTS 5 *
******************************************************************
|
À la fin du processus SCF, le programme CRYSTAL écrit des informations sur le système cristallin et sa fonction d'onde sous forme de données séquentielles non formatées sur le fichier fort.9, et sous forme de données formatées sur le fichier fort.98. Les données formatées permettent le transfert d'une plate-forme à l'autre.
Les propriétés à un électron et l'analyse de la fonction d'onde peuvent être calculées à partir de la fonction d'onde SCF en exécutant le programme properties. La séction propriété doir se terminer par le mot clé END.
Les données de la fonction d'onde sont lues (lorsque fort.98 est utilisé, le premier mot-clé doit être RDFMWF, pour convertir les données formatées en données binaires) lors de l'exécution des propriétés, et ne sont pas modifiées.
Les données incluent :
Les vecteurs propres hamiltoniens ne sont pas stockés, ils doivent être recalculés si nécessaire (mot clé NEWK).
Le but de ce didacticiel est d'apprendre à extraire certaines propriétés de base à un électron de la fonction d'onde :
Les cartes de densité de charge électronique totale (mot-clé ECHG) fournissent une représentation graphique de la distribution électronique totale.
La densité de charge est calculée dans une grille de points.
Des informations utiles sont obtenues en considérant des cartes de différence, données comme une différence entre la densité électronique du cristal et une densité électronique "de référence". La densité "de référence" peut être une superposition de distributions de charges atomiques (ou ioniques). Dans ce cas, deux exécutions identiques de l'option ECHG doivent être soumises (dans le même fichier d'entrée) et la seconde doit être précédée du mot-clé PATO, pour calculer une matrice de densité comme superposition des densités d'atomes).
La densité de charge aux points de grille est enregistrée dans le fichier input_filename.f25.
En général, vous devez suivre le schéma indiqué dans le tableau qui fait référence à MgO :
| ECHG | keyword | |
| 0 | order of the derivatives: if not 0, the charge density gradients are computed | |
| 65 | number of point along the B-A segment (see CRYSTAL User's Manual) | |
| COORDINA | cartesian coordinates of points A, B, C defining the window in a plane | |
| -4. -4. 0.0 | cartesian coordinates of point A (Angstrom) | |
| 4. -4. 0.0 | cartesian coordinates of point B (Angstrom) | |
| 4. 4. 0.0 | cartesian coordinates of point C (Angstrom) | |
| MARGINS | margins are added to the window, in the order AB,CD,AD,BC | |
| 1.5 1.5 1.5 1.5 | width of the margins, in the order AB,CD,AD,BC | |
| END |
Exercice :
ECHG 0 65 COORDINA -4. -4. 0.0 4. -4. 0.0 4. 4. 0.0 MARGINS 1.5 1.5 1.5 1.5 END PATO 0 0 ECHG 0 65 COORDINA -4. -4. 0.0 4. -4. 0.0 4. 4. 0.0 MARGINS 1.5 1.5 1.5 1.5 END END |
A | | | | B___________________C The position of the three points is given by their cartesian coordinates |
2) Utilisez XCrySDen pour visualiser la carte de densité de charge électronique totale et la carte de densité électronique différentielle, utilisez xcrusden :
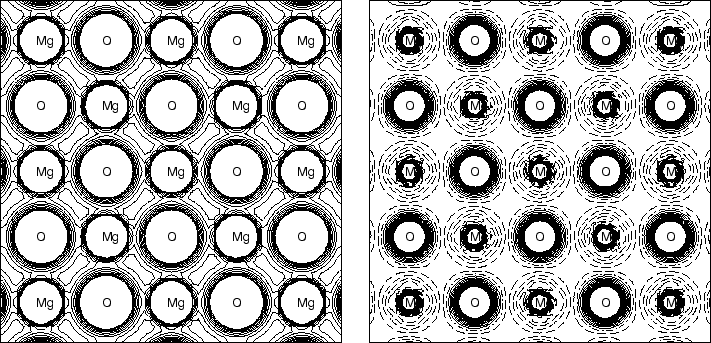
MgO electron density: total and difference maps (linear scale).
Des informations utiles peuvent être extraites du spectre des valeurs propres de l'hamiltonien (HF ou DFT) : la structure de bande etc…. La topologie des bandes occupées et des premières bandes de conduction sont généralement correcte. La forme de la surface de Fermi dans les métaux peut donc être prédite avec une assez grande précision.
Dans le fichier d'entrée de la structure des bandes certaines informations doivent être spécifiées :
1. Le chemin dans la zone de Brillouin de l'espace réciproque. La forme de la zone de Brillouin dépend du réseau réciproque, qui est univoquement défini par le réseau direct (c'est-à-dire le réseau de Bravais du système).
2. La plage de bandes à écrire en fort.25 pour le traçage. Le calcul des bandes peut être très exigeant pour les grands systèmes.
Afin de calculer les bandes, les données d'entrée suivantes sont requises :
1. Le nombre de segments définissant le chemin dans l'espace réciproque ;
2. Le facteur de rétrécissement en terme duquel sont exprimées les coordonnées de l'extrémité des segments ;
3. Le nombre total de k points le long du chemin ;
4. La première bande à enregistrer ;
5. La dernière bande à enregistrer ;
6. L'option de traçage (1 écrit les données sur l'unité fortran 25);
7. Les options d'impression (0 pour aucune impression).
Exercice :
Considérons 5 segments le long du chemin suivant (voir figure ci-dessous) :
Gamma(0,0,0) --> X(1/2,0,1/2) --> W(1/2,1/4,3/4) --> L(1/2,1/2,1/2) --> Gamma(0,0,0) --> W(1/2,1/4,3/4)
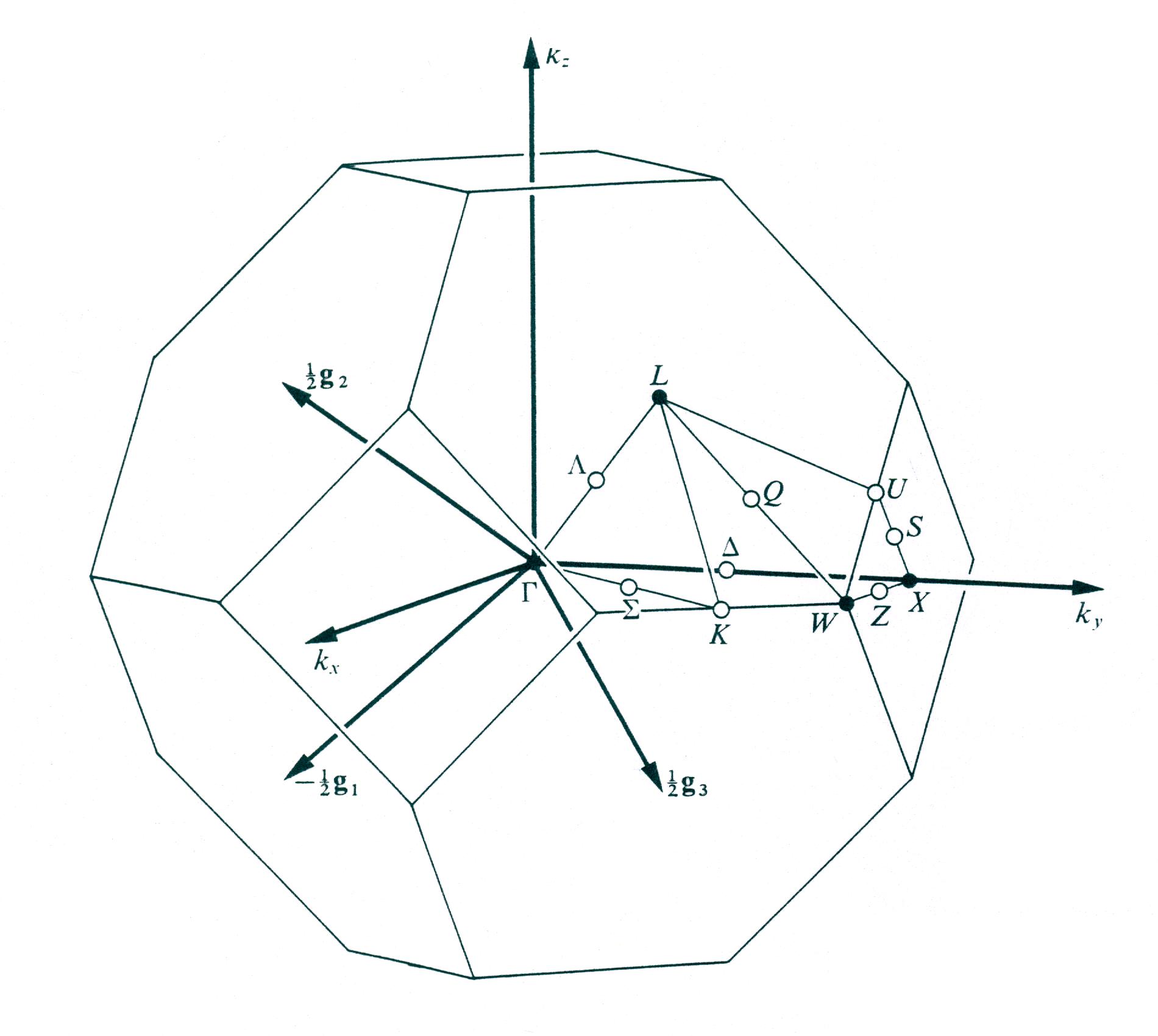
The Brillouin zone for the face centered cubic lattice.
En utilisant 12 comme facteur de rétrécissement, nous obtenons les coordonnées des points de réseau spéciaux ci-dessus rapportés dans le pont d'entrée commenté ci-dessous.
Par exemple, les trois entiers 6 3 9 du tableau correspondent au point W de coordonnées (6/12 3/12 9/12), ou de manière équivalente (1/2 1/4 3/4) dans l'espace réciproque.
Nous voulons tracer TOUTES les bandes, c'est-à-dire que nous devons considérer TOUTES les AO (voir la sortie SCF obtenue précédemment). Voici rapporté et commenté le fichier d’entrée pour calculer la structure de la bande de MgO :
| BAND | keyword |
| MgO | title |
| 5 12 30 1 18 1 0 |
5: number of segments in the reciprocal space to explore; 12: shrinking factor in term of which the coordinates of the extreme of the segments are expressed; 30: total number of k points along the path; 1: first band to be saved; 18: last band to be saved; 1: plotting option (if 1, write data on fortran unit 25); 0: no printing option are activated |
| 0 0 0 6 0 6 |
segment in the reciprocal space: Γ |
| 6 0 6 6 3 9 |
X |
| 6 3 9 6 6 6 |
W |
| 6 6 6 0 0 0 |
L |
| 0 0 0 6 3 9 |
Γ |
1) Exécutez un calcul de propriétés avec l'entrée suivante (mgo_band.d3) :
BAND MGO 5 12 30 1 18 1 0 0 0 0 6 0 6 6 0 6 6 3 9 6 3 9 6 6 6 6 6 6 0 0 0 0 0 0 6 3 9 END |
2) Utilisez XCrySDen pour visualiser la structure de bande.
Le calcul de la densité d'états nécessite les vecteurs propres de Fock/KS en les k points définis par le réseau de Pack-Monkhorst.
Le mot clé NEWK permet de calculer des vecteurs propres à partir d'une matrice hamiltonienne donnée.
Dans le fichier d'entrée pour le calcul de la densité d'états (DOSS) les informations suivantes doivent être spécifiées :
Dans le cas d'une densité projetée sur un sous-ensemble d'AO, d'autres données doivent être spécifiées dans le fichier d'entrée :
Exercice :
La densité d'états est projetée sur l'ensemble des AO de l'atome 1 (Mg) et de l'atome 2 (O). Fichier mgo_doss.d3 :
| NEWK | keyword |
| 0 0 |
0: shrinking factor for reciprocal space Pack-Monkhorst net is taken from the SCF input 0: shrinking factor for reciprocal space Gilat net is taken from the SCF input |
| 1 1 |
1: calculation of the density matrix from the new eigenvectors (it corresponds to one more SCF cycle) 1: print options |
| 66 100 |
66: printing option number 100: max number of eigenvalues printed |
| DOSS | keyword |
| 2 100 7 14 1 14 0 |
2: number of projections (the total DOS is always performed); 200: number of points along the energy axis in which the DOSS is calculated; 7: first (first valence band) 14: last band (fourth virtual band) 1: plot option (if 1, the program stores the data in file fort.25); 14: degree of the polynomial used for the DOSS expansion; 0: printing option |
| -1 1 | projection onto all the AOs (-1) of Mg (1, first atom) |
| -1 2 | projection onto all the AOs (-1) of O (2, first atom) |
1. Calculez le DOSS pour MgO, mgo_doss.d3 :
NEWK 6 6 1 1 66 100 DOSS 2 200 7 14 1 14 0 -1 1 -1 2 END |
2. Le fichier de sortie doit contenir :
###############################################################################
TOTAL AND PROJECTED DENSITY OF STATES - FOURIER LEGENDRE METHOD
FROM BAND 7 TO BAND 14 ENERGY RANGE -0.11613E+01 0.11754E+01
###############################################################################
NUMBER OF LEGENDRE POLYNOMIALS 14
NUMBER OF SYMMETRIZED PWS FOR EXPANSION 16
NUMBER OF K POINTS OF SECONDARY NET 16
NUMBER OF PROJECTIONS 2
NUMBER OF ENERGY POINTS 198
*** PROJECTION 1 ATOMIC ORBITALS 1 2 3 4 5 6 7 8 9
*** PROJECTION 2 ATOMIC ORBITALS 10 11 12 13 14 15 16 17 18
BAND INTEGRATED DOSS PER PROJECTION AND TOTAL
1 2 T
7 0.00991 1.99009 2.00000
8 0.01277 1.98723 2.00000
9 0.01147 1.98853 2.00000
10 0.00491 1.99509 2.00000
11 0.47259 1.52741 2.00000
12 1.03565 0.96435 2.00000
13 1.24271 0.75729 2.00000
14 1.44140 0.55860 2.00000
|
3. Utilisez XCrySDen pour visualiser la DOS.
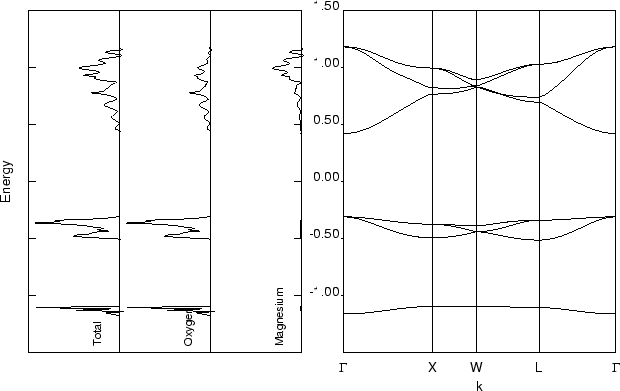
MgO doss and band structure.
Répétez le calcul proposé précédemment pour le MgO avec le Si et le Be. Comparez les résultats. Notez que Be est un solide métallique, l'énergie de Fermi doit être calculée avec une valeur du facteur de rétrécissement de Gilat, ISP = 2*IS (facteur de rétrécissement Pack-Monkhorst net).
